Plant Protection Products Impurity Assay by GC-FID with Hydrogen Carrier Gas
Introduction
This application note is part of a series discussing submission of a chemical product to regulatory bodies.
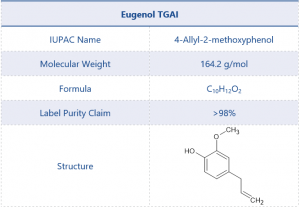
A commercially available sample of Eugenol was chosen as an example chemical product. Eugenol is a terpene which makes up around 80% of clove leaf oil and is attributed to the distinctive smell of cloves. Eugenol is also a powerful insecticide and when imported for this use falls under the requirements of the Plant Protection Products (PPP) or pesticide regulations.1
The chemical structure and other details of Eugenol can be found in Table 1.
In this application note we will look at EU and UK regulations for submission of technical active substances and PPP No 283/2013 (Annex Section 4) and 284/2013 (Annex Section 5). These regulations have a guidance document SANCO/3030/99 rev5 which sets out the testing required for a successful submission of a product.2
Plant protection products (PPP) or pesticides are subject to strict regulations for import to countries all over the world.
In the 1st application note we discussed how to successfully validate and assay for the active ingredient (AI) content (See AN164). The 2nd application note discussed how to determine which impurities require identification and quantification through an impurity screening process (see AN165).
In this application note we will validate and assay for the relevant impurity methyl eugenol and an example significant impurity 4-(prop-2-en-1-yl)phenol. For ease we will refer to methyl eugenol as impurity 1 and 4-(prop-2-en-1-yl)phenol as impurity 2 from here on.
Relevant impurities which are defined as being “impurities of toxicological and/or ecotoxicological or environmental concern”. These are defined by the product, for example eugenol, when submitted for use as a PPP, UK and EU regulations define methyl eugenol as a relevant impurity that can be present at a maximum of 0.1% of the technical material.3
Whereas significant impurities are impurities that are present at or above a % defined by the relevant regulation. So for example for PPP products in the EU/UK the SANCO guidance document dictates that any impurity >0.1%.
Determining impurity content is a requirement across many regulated industries from pharmaceutical, food, agriculture, and environmental.
Helium has long been the standard carrier gas for GC applications however, due to recent global shortages many companies are keen to find alternatives.4 The application described in this note has been performed using an alternative carrier gas, hydrogen. This can be produced with generators using renewable energy sources and so is a cleaner, greener option as well as avoiding the current issues over helium supply.
Table 1. Test Item Details
This application can be performed on either the SCION Instruments 8300 GC or 8500 GC platforms equipped with an 8400 Pro Autosampler, S/SL injector, and FID detector.
Experimental
Section 4.1.2. of the SANCO guidance document details the validation requirements of any method used to quantify the impurities of a product.
The document sets out strict requirements for specificity, linearity, recovery and precision of the method. The limit of quantification (LOQ) of each impurity must also be determined, this is established through successful recovery of fortified samples at the specified levels within the guidance document for significant impurities and at least at 20% less for relevant impurities. In this instance impurity 1 must be recovered at ≤0.08% and impurity 2 at ≤0.1%. The limits for recovery are defined in appendix 2 section 7.1 of the SANCO document.
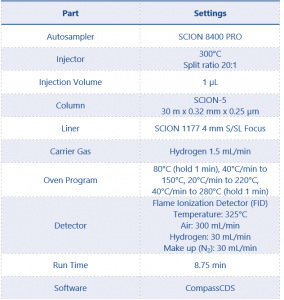
The instrument method parameters can be found in Table 2.
A suitable internal standard was chosen, hexadecane (purity 99.8%), which had a similar response factor to the target analytes and did not interfere with the active ingredient or any impurities present.
Table 2. Instrument Parameters
The internal standard solution was prepared by weighing 125mg hexadecane analytical standard into a 50mL volumetric flask, this was made to volume using Acetonitrile (HPLC grade 99.8%) and mixed well.
Linearity samples were prepared as per the guidance document at 5 concentrations ranging at least +/- 20% of the expected concentrations of the impurities. A stock solution was prepared by weighing 10mg of each impurity and making to volume with acetonitrile. A number of dilutions were then performed by pipetting 0.25, 0.5, 1, 2, 3, and 5mL into separate 10mL volumetric flasks. 1 mL of the internal standard solution was added to each sample which were then made to volume with acetonitrile.
Recovery samples were to be determined at 2 levels with the lower level determining the LOQ. A stock QC solution was prepared by weighing 10mg of each impurity into a 50mL volumetric flask and making to volume with acetonitrile. SANCO defines precision as requiring a minimum of 5 determinations, we chose to prepare 6 fortified samples at each level. 3 blank samples were also prepared to allow for standard addition method (subtraction of any impurity present in the technical material). These samples were prepared by weighing 100mg TGAI eugenol into separate 10mL volumetric flasks. 1mL internal standard solution was added to each sample. The 3 blank samples were then made to volume with acetonitrile.
The low level recovery samples had 0.4mL of the QC stock solution added, the high level recovery samples had 1.5mL QC stock solution added. All samples were then made to volume using acetonitrile.
For the calculation of the impurity levels present in the technical material each batch of eugenol being tested (in this case a single batch) had samples prepared in duplicate. 100mg eugenol was weighed into separate 10mL flasks, 1 mL internal standard solution was added and the samples made to volume with acetonitrile.
Results and Discussion
The instrument method stated in Table 2 gave excellent specificity. Figures 2 – 4 show example chromatograms of a blank (diluent only), linearity standard, and technical sample.
These chromatograms clearly demonstrate the specificity of the method, showing excellent resolution between all the components as well as good peak shape of both the impurities and the internal standard.
SANCO defines the criteria for acceptable linearity as a correlation coefficient (r2) value as >0.99. Tables 3 and 4 show the linearity results for impurities 1 and 2.
Table 3. Linearity results for impurity 1 (calculated using the peak area ratio of impurity 1/Internal standard)
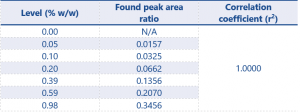
Table 4. Linearity results for impurity 2 (calculated using the peak area ratio of impurity 1/Internal standard)
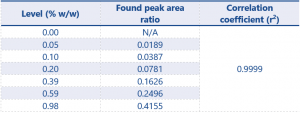
The r² value of 0.9997 demonstrates excellent linearity, greatly exceeding the criteria defined by the SANCO document.

Figure 2. Example Chromatogram of a blank sample (Acetonitrile only)
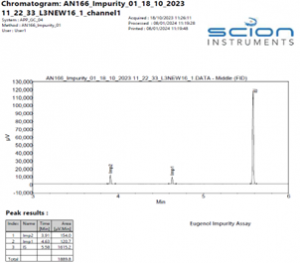
Figure 4. Example Chromatogram of an impurity linearity standard
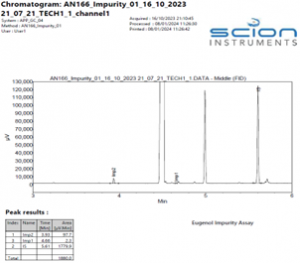
Figure 6. Example Chromatogram of a Eugenol TGAI sample
Appendix 2 of the SANCO guidance document details the criteria for validation results. For recovery any active substance or impurity present at ≥0.01 – <0.1% requires a mean recovery of 75 – 125% to be considered acceptable. For substances present at ≥0.1 – <1% a mean recovery of 80 – 120% is required.
Precision results are determined to be acceptable using the Horwitz equation. The Horwitz equation corresponds to an exponential relationship between the estimated intra-laboratory repeatability (RSDr) and concentration (c).
![]()
For example for the LOQ determination of both impurities which is performed at 0.08% w/w the %RSDr or Horwitz value will be 3.92.
With this obtained value for the Horwitz value the Horwitz ratio (Hr) can be calculated, using the obtained repeatability (%RSD) of the target component.
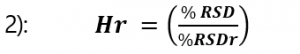
From this calculated value (Hr) the following criteria are applied:
Hr ≤ 1, acceptable.
1 < Hr ≤ 2, acceptable in case of a suggested explanation.
Hr > 2, not acceptable.
Assay accuracy and precision results are shown in Tables 5 – 8.
Table 5. Assay Accuracy and Precision results for impurity 1 at 0.08% w/w (LOQ determination)
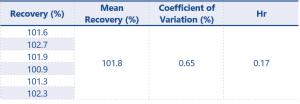
Table 6. Assay Accuracy and Precision results for impurity 1 at 0.30% w/w
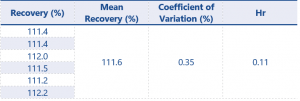
Table 7. Assay Accuracy and Precision results for impurity 2 at 0.08% w/w (LOQ determination)
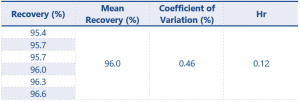
Table 8. Assay Accuracy and Precision results for impurity 2 at 0.29% w/w
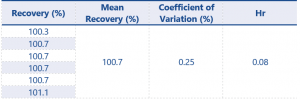
For results at the 0.08% w/w level recovery should be between 75 – 125%, and at the higher level recovery should be between 80 – 120% – these values have been greatly exceeded by the method.
The successful recovery of both impurities at the 0.08% w/w level now means the LOQ can be determined as being 0.08% w/w for each impurity, this fulfils the requirements of the SANCO document with regards to LOQ.
The results for assay accuracy and precision easily pass the criteria set by SANCO. With these results the impurity method can now be considered fully validated and the content of impurities 1 and 2 present in the product can be accurately quantified.
The results of the impurity assay can be found in Table 9.
Table 9. Results for impurity content
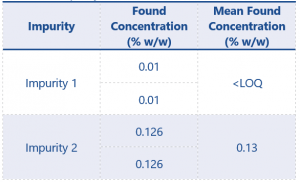
The 2 replicates of the eugenol product gave a mean found concentration of <LOQ for impurity 1. As a relevant impurity methyl eugenol has been defined as being allowed at a maximum content of 0.1%. The method in this application note has shown methyl eugenol to be present at <0.08% w/w and therefore this product would be considered acceptable in terms of methyl eugenol content for submission as a PPP within the UK or EU.
The 2 replicates of the eugenol product gave a mean found concentration of 0.13 % w/w for impurity 2. This impurity had been determined as a significant impurity during the screening process (see application note AN165). The method detailed in this application note has determined an LOQ at 0.08% w/w with respect to impurity 2 and has successfully quantified the impurity in line with the SANCO guidance document.
As stated during the introduction, these 2 impurities were chosen as example relevant and significant impurities. In reality this eugenol product would not be considered acceptable for submission as a PPP due to the % of impurities present which means this particular product does not meet the ≥99% eugenol content required by UK and EU guidelines (see application note AN165 for full details on all identified impurities).
This batch of eugenol would have been eliminated during the screening process and another batch meeting the criteria would have been chosen for submission as a PPP. However the testing in this application note does give a clear example of how an impurity content method would be fully validated and assayed for to meet EU and UK regulations.
Conclusion
Impurity content of a chemical product is a requirement of testing across several regulated industries. In this application note we have seen how to fully validate an assay for a relevant impurity (methyl eugenol) and a significant impurity (4-(prop-2-en-1-yl)phenol) in compliance with the guidance document SANCO/3030/99 rev.5 for submission of a plant protection product or pesticide within the EU or UK.
This testing would also form the basis for submission to multiple other countries such as the US, Mexico, Brazil, Canada, and Australia. SCION instruments recommends checking with local regulatory authorities to ensure all testing and reporting requirements are met, or contact the SCION applications team for assistance.
The principles used in this application note can be applied to not only insecticides, but to other environmental pesticidal products such as biocides and fungicides as well as across other industries such as cosmetics, food, and drug testing.
The product tested in this application note (eugenol) was shown to fall below it’s label claim of 98%. With the AI assay giving a purity value of 97.3%. The screening process confirmed the presence impurities totalling 2.79% which would give a closure value of 100.1% (SANCO defines acceptable closure of 98 – 102% w/w). The impurities assayed for in this application note were to give an example of how to assay for relevant and significant impurities – this eugenol product would not pass the requirements of import for use as a PPP, and would have been excluded during the screening process with a more suitable batch chosen for submission.
This product was imported under less stringent (REACH) regulations and was not intended for use as a PPP. However, this result highlights the importance of rigorous regulatory testing to ensure the quality of products being imported.
References
- Commission implementing regulation (EU) No 546/2013, Approval of Eugenol for use as a PPP, 14 June 2013, L 163/17
- European Commission, SANCO/3030/99 rev.5, 22 March 2019
- Commission implementing regulation (EU) No 546/2013, Approval of Eugenol for use as a PPP, 14 June 2013, Annex I
- Innovation News Network, Helium Shortage 4.0, Accessed January 2024, https://www.innovationnewsnetwork.com/helium-shortage-4-0-what-caused-it-and-when-will-it-end/29255/